
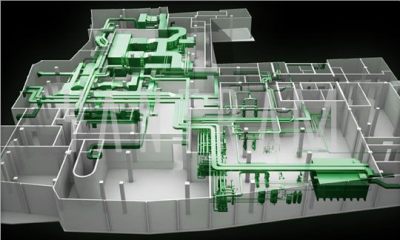
Any project is successful when the right team works on it. For any construction or renovation project an MEP consulting firm is just as important as an architect or a builder. MEP stands for Mechanical, Electrical and Plumbing; a type of engineering that focuses on creating a safe environment for human use.
There are various reasons why MEP consultants are highly important for any building project.
We’ve listed down the top ten reasons.
- Mechanical – For any building project, residential or commercial, heating and ventilation services are essential. This is where the MEP engineers step in. They are responsible for the installation and maintenance of air conditioners, heaters, exhaust and smoke control. An MEP consultant will ensure their designs provide comfort and run efficiently.
- Electrical – Electrical services are indispensable. Your project will be incomplete without electricity and lighting. MEP consultants are responsible not just for lighting but also other electrical services such as fire alarms, security systems, smoke alarms etc. They provide solutions to the best services that are energy efficient and environment friendly.
- Plumbing–The plumbing aspect focuses on fire suppression systems as well as gas delivery systems in medical and laboratory settings. They are also responsible for water and waste management systems such as drinking water, irrigation water, sewers etc.
- Effective design – MEP consultants provide the best design solutions that are cost effective and worthwhile in the long run. These designs should not only employ the most recent and competent technologies but also provide foresight to ensure maximum lifecycle.
- Environment friendly – Whether it is energy conservation or water conservation, the experts certainly know the best. These consultants aim at providing services that utilize environment friendly products in order to reduce excessive consumption of resources.
- Cost Efficient – Every project that you take up has a stipulated budget that needs to be followed. With the help of MEP engineers not only do you manage to stay inside budget but also end up with systems that will be cost efficient in the long run.
- Optimum use–The expertise of MEP engineersensures that the products and systems required for your project provide maximum utilization. They ensure that maintenance of systems is limited and simple and workers are well trained to handle issues, if any.
- . Indoor environment –While focusing on creating an ecofriendly environment for the outdoors, it is also vital to build a safe and healthy environment indoors. Indoor environment quality refers to the use of natural daylight, moisture control, optimizing systems etc. A healthy indoor environment equates a safer environment for everyone.
- Building mechanism –Every system is connected to centralized hardware and software networks that control the indoor and outdoor features in a building. For example, your construction project requires HVAC commissioning; MEP consultants will cover the entire procedure for the project. Automation systems are required to build and maintain a building’s optimal operational performance and ensure safety and comfort the occupants.
- Expertise- It is always better to leave the job in the hands of the one that knows the best. MEP consultants are highly trained and experienced in their field. They work closely with architects and owners to provide successful coordination of building systems.
Panorama offers a variety of services in the mechanical, electrical and plumbing divisions for pharmaceutical and chemical sectors. Our specialized services will help your company with the sustenance it requires. We are a leading MEP Consulting Firm in Mumbai with numerous successful projects.









Recent Comments